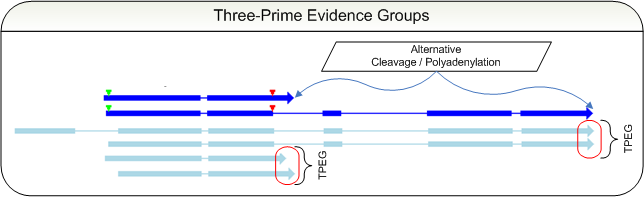
Three-prime Evidence Groups &
Alternative Cleavage / Polyadenylation Site Usage
Examples
Documents
Other Links
The analysis of transcriptional termination sites requires
evidence alignments related to such sites. In terms of the GAEVAL algorithm
the evidence currently used includes 'full-length' cDNA sequences and
'three-prime' EST sequences. In order to capture this information in the GFF
file input it is REQUIRED that these sequences include the attributes
'full_length_transcript=1' and 'end_type=T' respectively. For an example of
this format see example4.gff3
Determining the exact nucleotide (or nucleotide region) at which transcription concludes for a gene
has been an active field of study for over three decades. However, with the increased availability of genomic sequence,
and thus an inherent need for gene annotation, tools and methods for accurate prediction of the downstream extent
of a gene are in demand now more than ever. Herein we discuss current knowledge of the biological properties
of the cleavage / polyadenylation site as well as methods for locating an approximate region of termination given available EST and cDNA evidence.
The Biology
In eukaryotes, the common RNA-POL II transcribed gene undergoes the processes of maturation
(ie. capping, splicing, and polyadenylation) concurrent with transcription. While the exact mechanism which
terminates transcription is unproven, the common assumption is that a lack of processivity of the RNA-POL II
caused by dissociation of stabilizing elongation factors in concert with cleavage/polyadenylation allow termination
to occur at the next thermodynamically favorable location (Zhao, 1999). Thus the exact point of pre-mRNA transcript termination
may or may not be conserved. However the site of cleavage/poyladenylation is generally static as shown by mutation
studies which produce transcriptional run-off when this site is altered. While the extent of transcriptional excess
may have regulatory consequences, identification of the cleavage/polyadenylation site is sufficient to determine the
sequence extent of mature mRNA as related to the annotation of a given transcriptional unit.
Genomic Landmarks for Locating a CPS
Cleavage / Polyadenylation sites are largely difficult to predict based
on sequence motifs due to their short and often degenerate nature. 3' Expressed Sequence Tags (ESTs) however, provide
an empirical method for locating the CPS. By accurate spliced alignment of the 3' ESTs, cluster groups can be used to
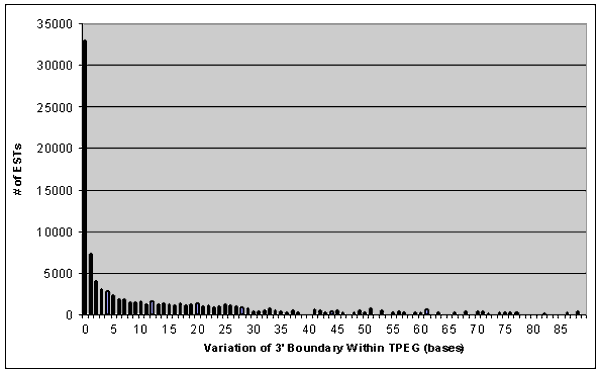
determine a localized region containing the CPS (Gautheret, 1998). For this analysis, 98,313 3' ESTs were clustered into 13,148 multi-member
clusters. Variation of the aligned 3' boundary within these Three-Prime EST Groups (TPEGs) is shown in figure 1.
As previously observed (Graber, 1999), multiple CPS signals closely spaced (on the order of tens of nucleotides)
result in variation of the 3' EST aligned ends (again on the order of tens of nucleotides). This analysis confirms
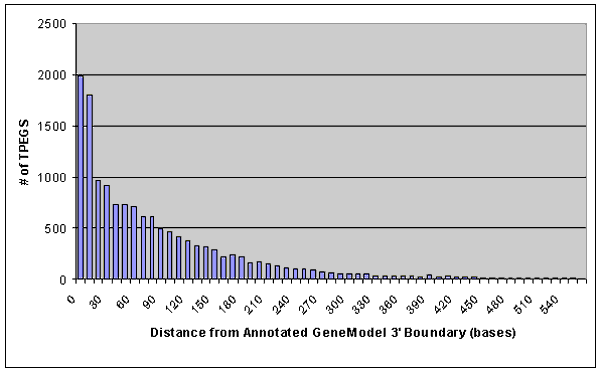
that over 95% of the current gene-model annotations containing TPEGs have 3' boundaries within 150 nucleotides of the TPEG defined
CPS (the vast majority have less than a 10 base difference). However exceptions do exists and have been flagged as
possible alternative polyadenylation sites, false gene extensions, and false intronic gene mergers.