

Community Annotation at PlantGDB
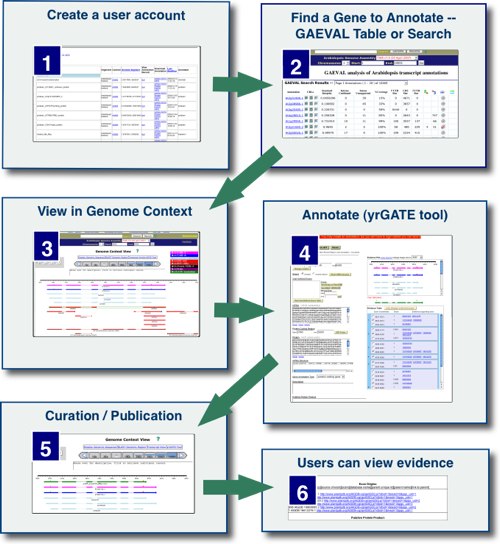
Overview
Annotation of a genome consists of creating gene models that define the most probable exon and intron structure(s), transcription start/stop(s), and other features for each gene that relate structure to function. A gene annotation may be based on spliced alignment of transcript evidence to the genome and/or computationally derived transcript structures. Accurate and complete gene models are essential to understanding genome biology. Community annotation harnesses the power of a research community to create and share gene annotations.
The Community Annotation system at PlantGDB makes it easy for users to annotate genome data for submission to a genome browser -- making it a true community database. Users can easily and quickly create and submit annotations via the yrGATE tool, a one-stop web interface for annotation. After a submitted annotation is approved by the PlantGDB administrator, it is incorporated into the genome browser and is publicly viewable on the yrGATE track. Submissions are reviewed weekly. Additionally, accepted annotations are added to our BLAST databases.
Annotating gene structure using yrGATE (Video Demo; 5 min 15 sec)
- Quicktime
- Flash
- (Download)
Key features of the Community Annotation system:
- A user-accessible community data site (Community Annotation Central)
- Genome Browsers for graphical display of spliced alignments (evidence) along with gene models
- A means to flag gene models that conflict with transcript evidence (GAEVAL Tables)
- An online community annotation tool (yrGATE)
- A curation process that displays approved gene models online.
- All annotation evidence can be viewed and retrieved.
The yrGATE* tool, the heart of the Community Annotation system, can be accessed directly through PlantGDB's genome browsers, which display current gene models and spliced alignment evidence for each species. Links to all xGDB genome browsers can be found in the xGDB Portal. Links to yrGATE examples and annotation case studies are found on the yrGATE Page .
* yrGATE: Your Gene structure Annotation Tool for Eukaryotes
This Help page provides a comprehensive guide to gene annotation at PlantGDB. To navigate to a topic, use the menu tree at left. In-text links to a related Help topic are green. Links to PlantGDB pages and external links are blue. For more information on annotation tools and methods, see references at the end of this document.
Quick Start
Annotate a Protein Coding Gene
- Log in or create an Community Annotation Central account from a Genome Browser Home Page (example: AtGDB).
- Return to the Genome Browser Home Page and enter the ID for a gene you want to annotate in the Search window (example: At1g01010.1). Then click 'Search: Genome'. The result is a Genome Context view of that gene.
- Open a new yrGATE window by clicking 'yrGATE Tool'. yrGATE displays gene models and evidence alignments along with tools for building a new annotation.
- Adjust genomic coordinates and transcription direction as needed.
- Build a gene model by choosing exons using the Evidence Table (radio button), the Evidence Plot (click on glyph), and/or the User Defined Exons window. The new gene model is displayed in green as "Your Structure", and the resulting sequence is displayed in the mRNA window.
- Validate your gene model using web portal
tools:
- Click ORF Finder and choose an open reading frame. The resulting protein sequence now appears in the Protein window.
- Optionally, run BLAST Query to determine similarity to known protein sequences.
- Enter Annotation Information about your
annotation as appropriate:
- Enter Gene Annotation Type (Protein coding gene, noncoding gene, etc.)
- Enter Gene Description, Putative Protein Product, Gene Aliases, Protein Aliases.
- Submit your annotation or Save it for later editing by clicking the appropriate button, and close the yrGATE window. Your annotation is now stored in your Community Annotation Central Account.
- PlantGDB will notify you by email when your annotation is approved or rejected. Once approved, your annotation and its evidence can be viewed in a Genome Browser or on Annotation Central.
Annotation Tutorials
Tutorials available at PlantGDB:
- Annotating gene structure using yrGATE (Video Demo; 5 min 15 sec)
- Annotation for Amateurs - Anyone can annotate genes. This site is designed to teach users the basics of gene annotation and provides access to several plant genomes which can be annotated. Once you learn to annotate genes you too can submit proposed annotations that will be evaluated by professionals. If your annotation is accepted it will be added to the appropriate genome database! Below are a list of questions and answers that explain more about this site and what you might find here.
- Gene Discovery and Annotation throught AtGDB - This module is intended for small groups or entire classrooms of science students in middle or high school. By working through the module, students will begin with understanding the structure of DNA and conclude with making gene annotations that will assist real scientists in research labs.
Community Annotation Central
The Community Annotation Central page
The starting point for annotation is the Community Annotation Central page for each genome. From here, previous user annotations can be viewed, submitted or deleted (if logged in). This web page also provides complete information about all curated community annotations, including links to Genome Context views and download options for FASTA or GFF format. Each Genome Browser has its own Community Annotation Central page (e.g. /AtGDB/CommunityCentral.pl). To access a page, click the Annotations@XxGDB link on the Genome Browser top menubar (see example below). Links to all Annotation Central browser pages can also be found at http://www.plantgdb.org/prj/yrGATE/.

Anyone can view existing annotations, but to contribute annotations you will need to register for a User Account (see below).
User Registration and Login
To acquire a User Account, click 'Register Here' on any PlantGDB home page, ('Register' in Genome Browser) or choose 'Sign up for an account' in Annotation Central. Note that although you can use the yrGATE tool without an account, you will not be able to save or submit your annotation. Username, password and an email address (to contact you once annotation is approved) are required in order to register.
Accessing Your Account
Once registered, you can login for session-wide access via any genome browser by clicking 'Login' and submitting username and password. The login window can also be accessed in the upper left header of any Genome Browser at PlantGDB.
After logging in, you arrive at the yrGATE: Community Annotation Central page from which you can view or download all previous annotations, or access your own annotations by selecting 'Annotation Account'. You can now annotate any XGDB browser using the same account; once logged in, your username appears in the upper left menu bar of each browser. The screenshot below shows the main features of the Community Annotation Central page.
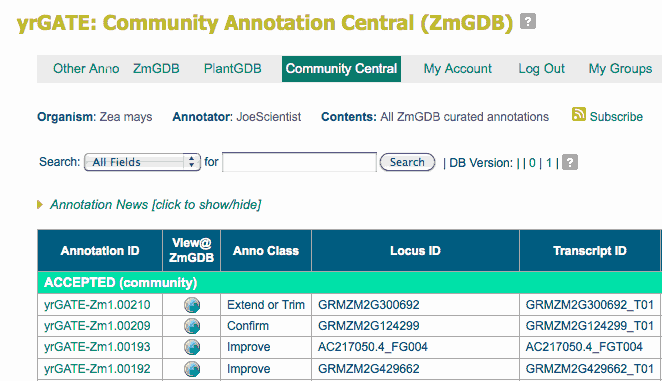
To create a new gene annotation, you will use the yrGATE tool, but first you need to locate a gene to annotate! The next section describes several ways to do this.
Finding a Gene to Annotate
The annotation process consists of building a gene model based on transcript or protein alignment evidence and/or computationally derived coordinates. For many genomes, computationally-derived gene models are displayed along with transcript evidence, and by comparing the two data sources, a more accurate gene model can be created.
To make this easier, the GAEVAL process flags genes with incongruence between evidence and computation and displays the results in the Genome Browser as a flag on each gene model track. A hover-over text box shows the number of introns confirmed by evidence alignments. The incongruent models are also listed in GAEVAL tables for each genome, which annotators can search for examples of potentially mis-annotated gene models.
(For genomes lacking gene models, annotators will need to rely on transcript/protein evidence alone or a combination of evidence and their own computations.)
Finally, Distributed Annotation Server (DAS) Data provides a means to display gene models and spliced alignments from other genome databases in a yrGATE window. The user can then create a Community Annotation based wholly or in part on DAS-served data.
In summary, locating a gene model to annotate can be accomplished in three ways:
- By searching or browsing within an xGDB genome browser (see below);
- By viewing pre-computed incongruent gene models using the GAEVAL tables (see below), or
- By displaying DAS-derived gene models (see below) in yrGATE.
Each of these options is detailed in the following sections.
From a Genome Browser
From the Menu Bar of any Genome Browser, you can select the ID for a gene you wish to annotate, or, alternatively, specify a genome segment (chromosome or BAC) and region in which to locate a gene.
- To specify a gene model or a splice-alinged transcript, enter the ID in the Quick Search box on the Genome Browser home page and click 'Search: Genome'.
- To access a region, type the genomic segment ID (chromosome, BAC gi, or scaffold number) and region (flanking base pairs) in the top menu bar and clicking 'Go'.

The Genome or Region search will open a Genome Context Page with all gene models and splice-aligned sequence represented as glyphs aligned below a genomic sequence track. See an example at AtGDB (Arabidopsis thaliana) showing a region of chromosome 1, from 1 to 10,001. The screenshot below shows a view of the same region with tracks identified. Notice a yrGATE annotation already exists for the gene model.
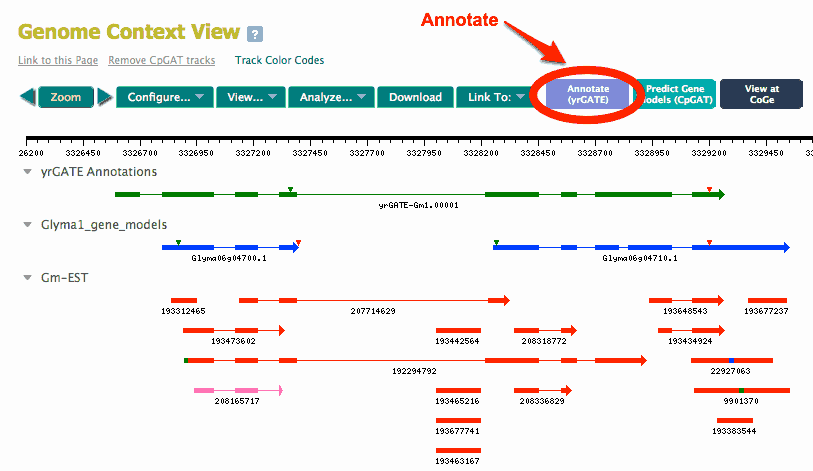
Compare the gene models (blue or magenta tracks) to the cDNA and EST spliced alignments for a locus. Do the gene models accurately describe of the splicing junctions indicated by transcript evidence, as well as transcription start and stop? To help answer this question, click the '+' in the Track Control for a gene model, and select "Show flags" radio button. The gene model glyph will
Through GAEVAL (Gene Annotation Evaluation) Tables
The GAEVAL tables at PlantGD provides a means to view all the known incongruent gene models for a genome, based on available spliced alignment evidence. GAEVAL pages are provided for all genome browsers at PlantGDB, and can be accessed from the left menubar of the GDB Home Page. For detailed information on GAEVAL, see GAEVAL Help section.
One useful feature of GAEVAL is the organization incongruent models by category:
- Alternative Splicing
- Alternative Transcript Termination
- Gene Fission
- Gene Fusion
- Erroneous Gene Overlap
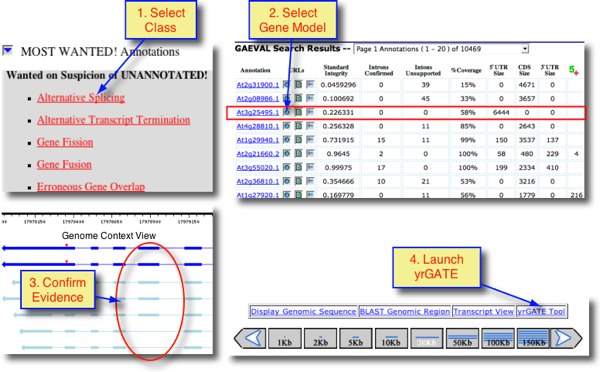
These categories are listed on the GAEVAL home page at, for example, http://www.plantgdb.org/XGDB/phplib/GAEVAL.php?GDB=At. Clicking a category opens a GAEVAL table with detailed information about each incongruent gene model and links to GAEVAL reports and Genome Context views. By clicking the Genome Context icon for a listed gene model in a GAEVAL table, it is possible to view the evidence alignments in the appropriate genome browser, and then open a yrGATE page to annotate the gene model. (See screenshot below.)
Through Distributed Annotation Service (DAS)
Distributed Annotation Server (DAS) Data provides a means to display gene models and spliced alignments from other genome databases in a genome browser. PlantGDB has implemented DAS for yrGATE in addition to a full-fledged DAS client service for xGDB browsers. yrGATE supports the DAS/1 standard described at http://biodas.org/wiki/DAS/1.
Using yrGATE to Annotate Genes
yrGATE (Your Gene Annotation Tool for Eukaryotes) is an online web tool for annotating genes, incorporating spliced alignment evidence from PlantGDB or other genome databases (see screenshot). yrGATE is available for all xGDB genome browsers, and registered users can submit annotations for curation and publication online (see for example green tracks in AtGDB). Detailed directions for using yrGATE are outlined in the following Help sections.
Launch yrGATE
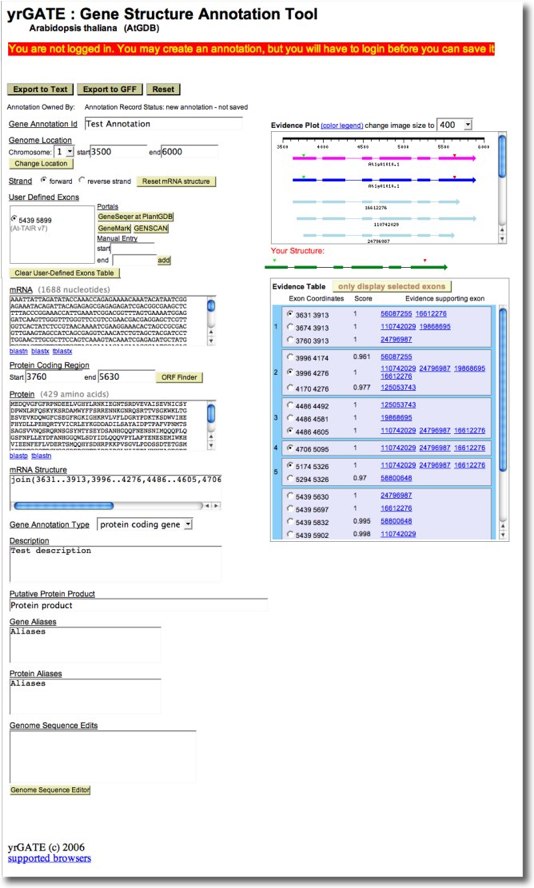
Let's assume you have identified a gene model to annotate, and have adjusted the genome coordinates to display all relevant evidence in a Genome Browser's Genome Context View (yrGATE is available for ALL Genome Browsers). Now, click on the yrGATE Tool link in an XGDB Browser (it's above the zoom/scroll toolbar on the right) to launch the yrGATE Gene Structure Annotation Tool. yrGATE provides an interactive form to assist your annotation submission. The screenshot below shows the yrGATE interface with a sample genomic region from the Arabidopsis thaliana genome browser AtGDB.
Now you are ready to apply the annotation tools in yrGATE. The yrGATE CA system provides flexible and dynamic mechanisms for swiftly and confidently entering a gene structure. Exons can be entered through several means:
- clicking on exons in the Evidence Plot (see below);
- selecting exons in the Evidence Table (see below);
- manually adding custom User Defined Exons (see below);
- Direct Entry of mRNA structure (see below)
- In addition, genomic sequence can be edited to correct errors
The following sections provide detailed instructions on how to enter a gene structure using yrGATE.
How to Use the Evidence plot
The Evidence Plot dislays in graphical all spliced alignments and gene models which meet quality thresholds. Color coding: gene models/mRNA (blue or magenta), proteins (black or brown), cDNAs (light blue) and ESTs (red). For a more detailed view, the window can be resized.
Clicking the exon structure in the Evidence Plot selects the exon and displays the current annotation gene structure with that exon as a green bar in "Your Structure". Clicking the ID beneath a glyph selects the entire gene structure. Introns are automatically defined by flanking exons. Clicking on an exon again, or on a similar exon in a different evidence sequence, will remove that exon from Your Structure. If an existing gene model (e.g. At1go1010.1) displays an exon that is distinct from any evidence-based exon and you believe it to be correct, you can click it to add it to the gene model. It then also be displayed in the User-Defined Exons Table.
Below is a screenshot of a typical Evidence Plot from yrGATE at AtGDB.
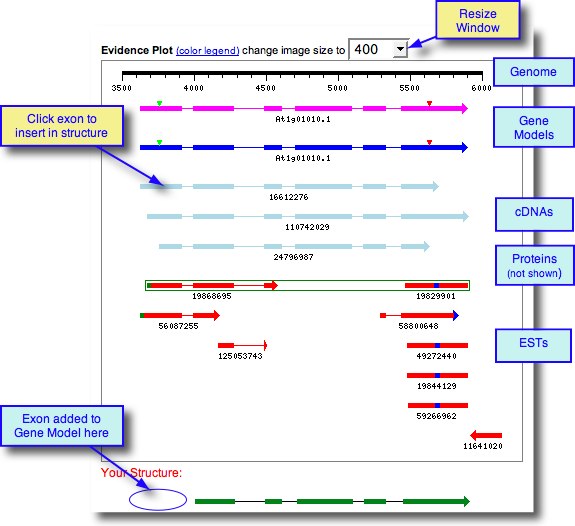
How to Use the Evidence Table
The Evidence Table displays all exons of EST and cDNA spliced alignments in the current genomic region, arranged in order from left to right. Exons are sorted by their left genomic coordinate and grouped into rows, which are mutually exclusive and represent the variants of an exon. Each row presents the coordinates, average GeneSeqer score, and EST and cDNA splice aligned sequences of the exon.
Click the radio button of the desired exon in the Evidence Table. Each evidence Sequence ID is hyperlinked to the GeneSeqer result files. The scores and Geneseqer result file allow a user to view the spliced alignment on the sequence level and to provide a confident annotation.
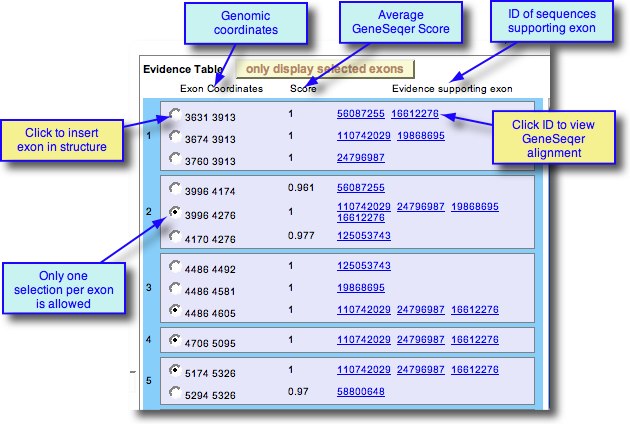
The mRNA Window
As exons are selected, the mRNA sequence is appended in the mRNA window. Blast portals are available to evaluate the sequences against GenBank nucleotide or protein.
Change Genomic Region/Strand
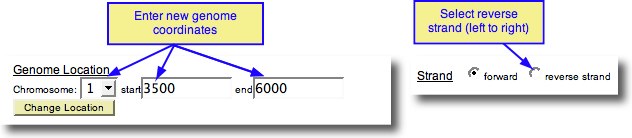
Only exons in the current genomic region are permitted to be entered through any of these mechanisms. To edit the range, under Genome Location, enter new start and end values, select a chromosome (if different), and click the Change Location Button. To change strand, select Reverse or Forward. NOTE: Use a range relatively close to the termini of your structure, so the resolution of the Graphic Plot and Your Structure displays all of the exons accurately, and the size of the Evidence Table is reasonable. Ranges larger than >20kb are not recommended.
Add User-Defined Exons
Manual Entry: The Manual Entry window allows user-defined exons (see screenshot below). To enter an exon not represented in the sequence evidence, enter 5' and 3' coordinates for your exon under Manual Entry and click 'add'. User defined exons are displayed in the larger box.To remove User Defined Exons, click on the 'Clear User Defined Exons' button, or de-select an individual exon radio button.
Portals: yrGATE includes several portals to ab initio gene prediction programs (see screenshot below). These buttons open a new window with results of the program for the current range of genomic sequence, with a button beside each predicted exon. When a button is clicked, the exon is added to your annotation. If the coordinates are shared with an evidence supported exon, the exon in the Evidence Table is selected. If it is not supported by evidence, it is added to User Defined Exons. When you are done with the portal, close the window. Note: It is recommended that annotations have evidence-supported exons to ensure inclusion in the genome database.

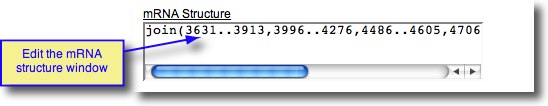
Direct Entry of mRNA structure
As a final alternative, the mRNA structure can be entered directly, or edited, by typing in the mRNA Structure window. See example screenshot below.
Reset mRNA Structure or yrGATE window
Click 'Reset mRNA' structure to start over with a gene model. To reset all fields, click the 'reset' button found at the top of the yrGATE window.
Note on Priority of Exon Entry Mechanisms
The following order of exon entry/removal mechanisms reflects their general priority in descending order:
- Reset mRNA structure button
- mRNA structure text entry = Evidence Plot Structure Selection
- Evidence Table button selection = Evidence Plot Exon Selection = User Defined Exon Entry
- Clear user defined exons button.
Tips
- Changing the strand will update the mRNA structure and Annotation Struture Preview
- Use a range relatively close to the termini of your structure, so the resolution of the Graphic Plot and Annotation Preview Structure displays all of the exons accurately, and the size of the Evidence Table is reasonable. Ranges larger than >20kb are not recommended.
- The mRNA and Protein Fields are updated anytime the structure is updated. Blast links can be used to blast the current structure.
Completing and Submitting Annotation
Enter Protein Coding Region (ORF)
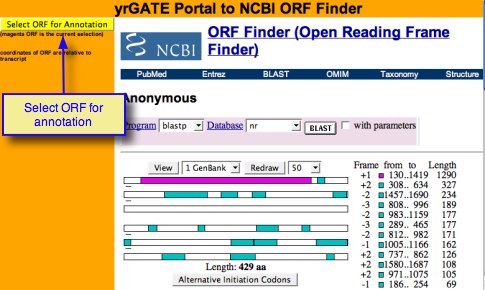
- After entering a gene structure, click on Open Reading Frame Finder Portal (ORF Finder). A new window, containing the Portal to NCBI ORF Finder, will open.
- Toggle between different open reading frames by clicking the rectangles. ORF Size, translated sequence, and location in the transcript are displayed.
- After selecting your open reading frame, (the selection is the magenta ORF) click on the button 'Select ORF for Annotation' . This will translate the transcript coordinates to genomic coordinates, paste them in the appropriate fields, display the arrows on the Annotation Structure Preview, update the mRNA and Protein preview fields, and close the ORF Finder Portal window.
Edit Genomic Sequence
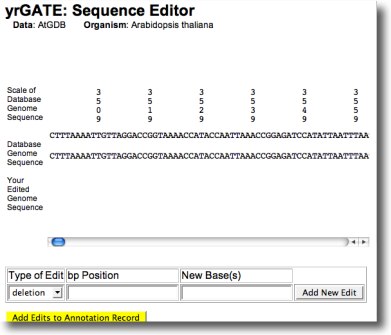
If an error in genomic sequence prevents accurate annotation, click 'Genome Sequence Editor' (bottom of yrGATE window) to open an editor window with a scrolling sequence track and and edit track. Enter base pair position, type of edit (deletion, change, insertion), and new base(s), then click 'Add Edits to Annotation' or 'Add Edit' to add another edit.
Enter Annotation Description Fields
- Enter an Annotation/LOCUS ID. Suggested format, yrGATE_At#g12345.1, where At = genus/species, # = chromosome number or BAC ID, 12345 = chosen identifier, '.1" is splicing variant number (if more than one). Example: yrGATE_At5g09920.1
- Enter Gene Aliases. (optional) Example: AL982373 AC008372
- Enter a Description. (optional) Descriptions typically consist of a functional assignment, source of genomic material, or source of annotation. Example:contains zinc finger domain;supported by cDNA 21404796
- Enter a putative protein product. After selecting, click on the 'blastn' or 'blastp' links. This pastes the appropriate sequence into NCBI's blast form. If satisfied with high scoring results, use this annotation information for your putative protein product field. Example: similar to exonuclease AY497556.
Save or Submit Annotation
If satisfied with the current annotation, click on the 'Submit' button at the top of the page. You will receive an email confirming that your submission has been received. Alternatively, to save an annotation for future editing, click on the 'Save' button. This annotation is now accessible from your account on the Annotation Central page and will be available for you to edit at a later time. See Editing your Annotation below.
Curation of yrGATE annotations
PlantGDB staff will review yrGATE submissions to Community Annotation and respond within 7 days with an email to you indicating whether or not your annotation has been approved. If approved, it will be displayed on the appropriate GDB web page (see Viewing in a Genome Browser. If not approved, the message will explain the reasons for rejection. If you want to try again, you can either edit and re-submit the existing annotation or start over. To send an email to PlantGDB Curation staff, click the 'Email' link in Community Annotation Central.
Viewing/Editing Annotations
Editing Your Annotation
Saved or submitted annotations are stored and can be viewed /edited at any time by the annotator. To access your annotations,
- First make sure you are logged in;
- Navigate to the genome browser for which you want to view annotations (e.g. ZmGDB).
- Click top menubar link 'Annotations@XxGDB' to access Community Annotation Central. From here you can view all publicly curated annotations. For more information see My Account section above
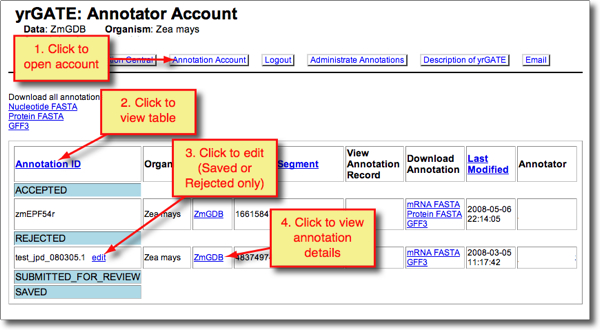
- To view your own yrGATE annotations (Accepted, Rejected, Submitted for Review, Saved), click 'Annotation Account' (see screenshot below).
- To continue editing a Saved or Rejected annotation, click 'edit' next to the annotation ID. This will launch a yrGATE window.
Viewing in a Genome Browser
If your annotations are accepted at PlantGDB, they will be displayed as a track in the Genome Context View of the genome browser. You can search for and view an annotation using its annotation ID or genomic location:
- Enter the annotation ID in the xGDB Search
window and click 'Genome' to view the
yrGATE gene model in its genomic context.
- For example, paste the ID 'yrGATE-At1g03905.1' in the Search window at AtGDB and click 'Genome'.
- (Alternatively, enter the genome segment and coordinates and click 'Go'.)
- The result is a Genome Context view of the genome with the yrGATE annotation shown as a green glyph in the center of the view.
- Click on the yrGATE glyph to view annotation details.
- (Alternatively, at step 1 click 'Records' instead of 'Genome', to jump immediately to the annotation details.)
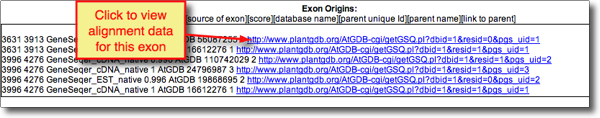
- When viewing annotation details, you can inspect the spliced alignment data for each exon by clicking on hyperlinked information under 'Exon Origins'.
- The resulting screen shows the spliced alignment evidence used and its origin: GeneSeqer, user input or other. See screenshot below.
Examples
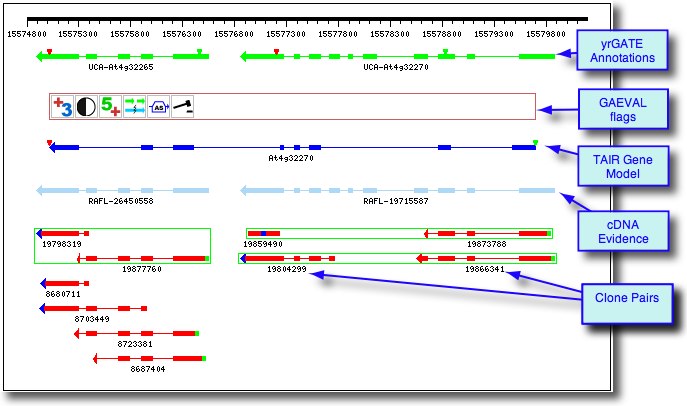
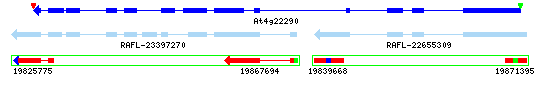
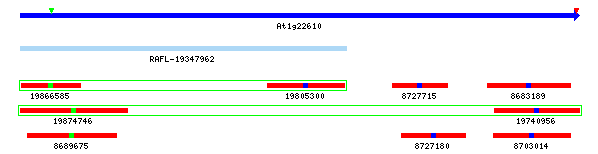
Example 1: Gene Model Split
At4g32265 / At4g32270: The yrGATE Annotations (green gene structures) in the figure below show how a user of AtGDB has provided an updated annotation. These annotations explain the inconsistency of the current gene model (At4g32270) with the spliced alignment of ESTs and cDNAs in this region. This model has also been flagged by GAEVAL link. By splitting this Gene Model into two annotations, the absence of overlap between the two full-length cDNAs (gi 26450558 & gi 19715587) is explained. In addition to the support gained by the alignment of these cDNAs, this yrGATE model is supported and consistent with the presence of three EST clone pairs (represented by the green box around a pair of ESTs). Via GeneSeqer at AtGDB, spliced alignment of the sequences in this region were threaded into two reasonable gene structures.
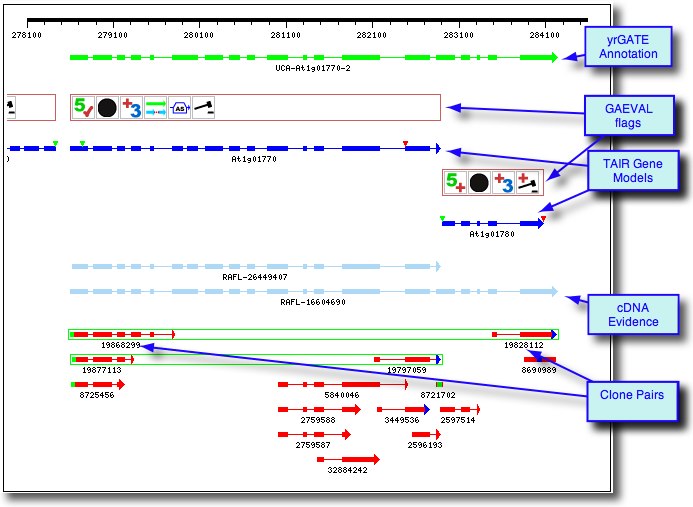
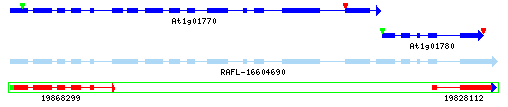
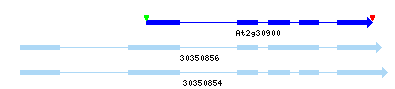
Example 2: Gene Model Merge
At1g01770-2: The yrGATE Annotation (green gene structure) in the figure below shows how a user of AtGDB has provided an updated annotation. This annotation explains the inconsistancy of the current gene model (At1g01780) with the spliced alignment of ESTs and cDNAs in this region. In addition to the support gained by the alignment of a full-length cDNA (gi 16604690), this annotation is supported and consistent with the presence of an EST clone pair (represented by the green box around ESTs 19868299 and 19828112). Via GeneSeqer at AtGDB, spliced alignments of the sequences in this region were threaded into a reasonable gene structure which suggests that Gene Model At1g01780 should actually be considered an extended isoform of Gene Model At1g01770.
What is GAEVAL?
GAEVAL (Gene Annotation Evaluation) is set of data summarizing inocongruence between gene models and evidence alignments for a given genome. The data are presented online in a variety of formats as described below. Symbols or glyphs are used to indicate the type of incongruence (e.g. alternate splicing) and the weight of evidence (credence) in support of the model or alternative model, and a numberic "Standard Integrity" score provides an overall assessment of how close the evidence matches the model. GAEVAL information is valuable for community annotation as it allows annotators to assess and prioritize the annotation needs for a genome.See screenshot below.

GAEVAL is available for any genome (xGDB) for which gene models are available, and it can be accessed in several ways:
- GAEVAL flags that appear on gene model glyphs in a GDB Browser, providing a visual cue as to which gene models are in need of fixing.
- GAEVAL Summary data in the Annotation Record page for each gene model.
- GAEVAL Tables with incongruences categorized by type, and quantitaive scores applied to each gene model. See also GAEVAL Types below.
The following section describes how to access and use GAEVAL tables as a source of genes to annotate.
GAEVAL Tables
GAEVAL tables are accessed from the left side menubar of each GDB. Click the link 'Problematic Gene Models' to display the page, 'GAEVAL analysis of transcript annotations'. This is a list of all the gene models for which transcript evidence does not fully support the model. The user has several options:
- Retrieve all annotations in a paged onscreen list (click 'Retrieve Annotations' button); or
- view by category under 'Wanted on Suspicion of UNANNOTATED'. (click a category: Alternative Splicing, Alternative Transcript Termination, Gene Fission, Gene Fusion, or Erroneous Gene Overlap
- View 'Best of Show', which are
annotations already submitted to yrGATE:
- Alternative Splicing
- Alternative Transcript Termination
- Gene Fission
- Gene Fusion
- User Annotated, Documented Isoforms, or "The Freaks" with unusual properties.
In addition, filters and sort order flags can be set for the retrieved data by setting check boxes and entering values for: Annotation Integrity, ANnotation Support Filters: Intron, Sequence Coverage or Boundary Coverage; Annotation Property, or Annotation Incongruence
The GAEVAL Table displays all gene models, filtered and sorted as selected by the user. The number of rows per window can be adjusted between 50 and 1000 records using the drowdown window at bottom of the table (default is 50).
Each row provides a gene model ID and at-a-glance information about the gene model. The URL icons in column 2 provide links to other views of each gene model (Genome Context view, Annotation Record, and GAEVAL report), and the other columns provide details about the gene model quality. Refer the to GAEVAL Table Key below for explanation of each column heading and icon.
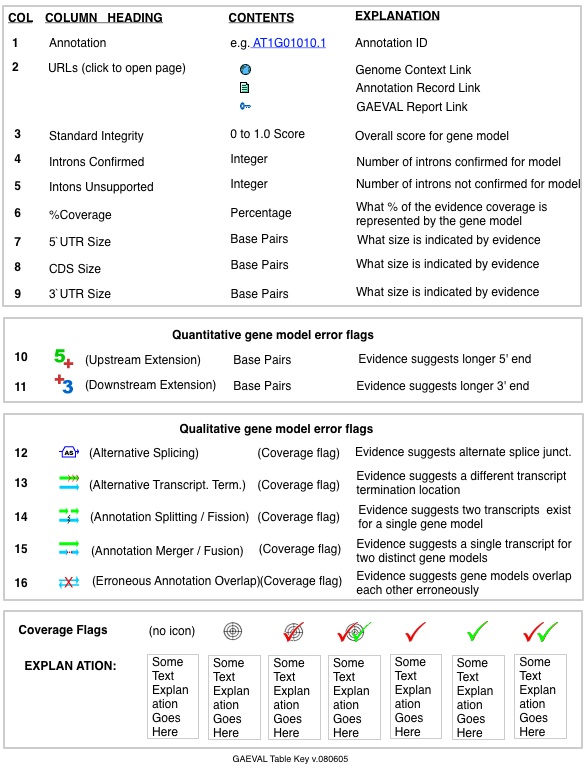
GAEVAL Report
Clicking on the "key" icon under URLs in the GAEVAL table opens a detailed report containing genomic coordinates for all incongruencies in the gene model. See figure below. The GAEVAL report can also be accessed from Genome Context view, by clicking on a GAEVAL flag associated with a gene model, or by viewing the Annotation Record for any gene model (click the "Full Report" icon).
GAEVAL Examples
The following examples illustrate several annotation scenarios and explain how the GAEVAL flags exemplify them. For more details on GAEVAL flags, refer also to the GAEVAL Types subsection below.
EXAMPLE 1 (see figure below). Here, a soybean gene annotation is presented along with cDNA and EST spliced aligments to the same region ("evidence"). The gene model predicts three introns, but cDNA and EST evidence contradicts the splicing model and the GAEVAL table shows this, as follows:
- Of the three predicted introns, cDNA and EST evidence supports only two of them, and the second intron is actively contradicted by cDNA transcript evidence. Instead, an apparent case of exon skipping has merged two introns into This scenario results in an "Alternate Splicing" with counterevidence check mark (red) in column 12 of the GAEVAL table.
- Note thatThe predicted start/stop locations are both contradicted.
Each row provides a gene model ID and at-a-glance information about the gene model. The icons in column 2 provide links to various views of each gene model, and the other columns provide details about the gene model quality. Refer the to GAEVAL Table Key below for explanation of each column heading and icon.
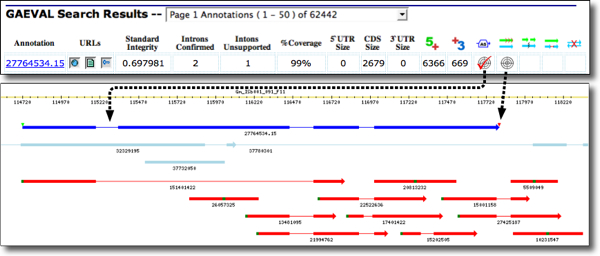
EXAMPLE 2 (see figure below). Here, the GAEVAL table shows an Arabidopsis locus, ATG3G275300.1 with multiple flags for incongruence (a). Clicking on the Genome Context URL displays evidence alignments below the gene model (which is in dark blue) (b). The GAEVAL flags and their hover-over text are indicated in (c). The flags denote: support for only four of nine introns; evidence of alternative splicing; alternate transcript termination (not surprising since the gene model does not include UTR regions); and also the possiblity of two distinct transcripts instead of the one indicated by the gene model. Clicking on any icon opens the GAEVAL report with genome coordinates associated with each incongruency.
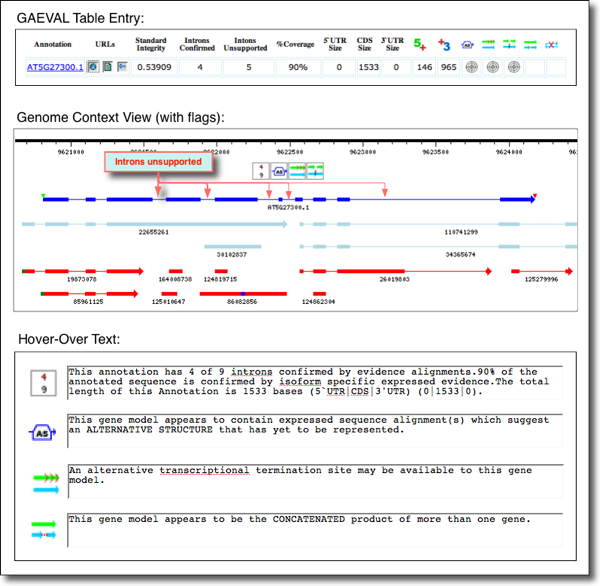
GAEVAL Rules and Symbols
The goal of the GAEVAL project is to assign quality scores to gene structure predictions and to note exceptional cases of incongruence of the annotation with experimental evidence. These scores can be used as a quick reference of the current accuracy of a given gene annotation and to prioritize the re-evaluation of incongruent gene predictions.
Finding the Cognate Locus
Evaluation of a gene prediction is achieved through comparison with known expressed sequences. This involves determination of the cognate locus (genomic origin) of the expressed sequences, correlating the sequences with a specific transcriptional isoform, and evaluating each annotation for incongruence.
Cognate loci were identified by spliced alignment of Arabidopsis EST and cDNA onto the Arabidopsis genome. The inherent quality of a cognate alignment is generally sufficient to distinguish it from alignment to homologous loci.
Details of these alignments are described in part in the manuscript by Zhu et al. (2003). See also supplementary information on the AtGDB Projects page
Assignment of a specific isoform
In many cases, assignment of an expressed sequence alignment to a specific gene annotation is trivial in that the alignment overlaps only a single annotation. However overlapping gene annotations, alternative splicing, clone-paired ESTs, and expressed sequences overlapping multiple adjacent annotations complicate matters. With this in mind, a scoring routine was devised as described in the Table below to ascertain the best assignment of an expressed sequence to a gene annotation isoform.
Table: Expressed Sequence Origin: Assessment and Assignment
| Score | Confirmation1 | Congruence2 | Containment3 | Consistency4 |
|---|---|---|---|---|
| 16 | + | + | + | + |
| 15 | + | + | + | - |
| 14 | + | + | - | + |
| 13 | + | + | - | - |
| 12 | + | - | + | + |
| 11 | + | - | + | - |
| 10 | + | - | - | + |
| 9 | + | - | - | - |
| 8 | - | + | + | + |
| 7 | - | + | + | - |
| 6 | - | + | - | + |
| 5 | - | + | - | - |
| 4 | - | - | + | + |
| 3 | - | - | + | - |
| 2 | - | - | - | + |
| 1 | - | - | - | - |
| 0 | No Overlap With Annotation | |||
- The expressed sequence spliced alignment confirms at least one splice site.
- Annotated splice junctions are completely consistent with the expressed sequence spliced alignment.
- The expressed sequence spliced alignment is completely contained within the annotation region.
- The expressed sequence spliced alignment does not contain any non-annotated splice sites.
GAEVAL Types and Quality Scores
Each gene prediction is now compared with its cognate, isoform-specific expressed sequence spliced alignments. This comparison determines support for predicted splice junctions, the extent of the annotation region, presence of alternative splice junctions, and alignment incongruence. Each of these properties is then scored, flagged, and listed. Methods for scoring and descriptions of each property are discussed below.
 Alternative
Splicing
Alternative
Splicing

Alternative splicing is determined through recognition of mutually exclusive incongruent exon designations (exon structures determined by spliced alignment which overlap annotated exons yet do not share common acceptor/donor boundaries).
Quality scoring: These events are evaluated by the number of spliced alignments confirming an alternative structure summed over all structures alternative to the given annotation.
 Gene
Annotation Fusion
Gene
Annotation Fusion
Annotation Fusion is necessary when gene predictions are recognized as incorrectly splitting a single gene into multiple adjacent gene annotations. This is made apparent by alignment of overlapping ESTs, clone-paired ESTs, and full-length cDNAs.
Quality scoring: These events are scored by evaluating the overlap of the isoform-specific ESTs or cDNAs with respect to the individual annotations. A score is then assigned based on the percentage of each annotation accounted for by the isoform-specific ESTs or cDNAs.
 Gene
Annotation Fission
Gene
Annotation Fission
Annotation Fission is necessary when gene predictions are recognized as falsely merging multiple adjacent gene structures into a single gene annotation. This can be determined through non-overlapping alignments of full-length cDNAs and the evaluation of 3' EST clusters (see a discussion of termination for more details). Cases in this category include mistaken classifications of alternative transcriptional termination sites. However, measure has been taken to classify alternative termination events as such when reasonable evidence is available to warrant such classification (see below).
Quality scoring: These events are scored based on the number of individually confirmed gene predictions determined to reside within the current annotation.
 Alternative
Transcript Termination Site
Alternative
Transcript Termination Site
Alternative transcriptional termination sites are often mistaken for incorrect gene annotation. Alternative termination may be a biologicaly relavent event and is currently the topic of many studies. The alternative termination events flagged by GAEVAL correspond to annotations which match the criteria for fission yet would also show evidence of fusion should independent fission products be evaluated. In other words, if cDNA or 3' cluster evidence exists that a gene could be terminated at both its annotated terminus and a secondary terminus, it is placed in this category.
Quality scoring: These events are scored relative to the maximum distance between any two hypothetical 3' termini.

 Upstream
Boundary Extension
Upstream
Boundary Extension
Underextended gene annotations represent gene predictions which do not make use of cognate, isoform-specific sequence alignments to determine the longest possible transcription unit.
Quality scoring: These events are ranked according to the size of the region of extended support.
Evidence Flags: Confidence in Predicted Gene Structure
The seven "Evidence" flags in columns 12-16 of the GAEVAL table indicate the degree of support for an alternative gene structure prediction. No icon means no evidence for an alternative structure. The presence of a target icon denotes evidence for an alternative structure, and check marks (with or without a target) indicate the degree and type of confirmation (red = Gene model-based; green = yrGATE annotation support) for that structure. Detailed descriptions of each flag are below:
(no icon). If there is no icon in a GAEVAL table column, then there is no evidence to support the type of incongruence indicated. For alternative structure (AS) type, lack of evidence could be for two reasons: 1) absence of EST or cDNA alignments to support or refute the model; or 2) absence of any predicted intronic sequence in the gene model and/or EST/cDNA at the locus.
Annotation action is warranted for the following types:
 Target:
Transcript evidence is present suggesting that
an alternative gene structure exists, of the
type indicated by the GAEVAL image in the
column heading.
Target:
Transcript evidence is present suggesting that
an alternative gene structure exists, of the
type indicated by the GAEVAL image in the
column heading.
 Target with
red check mark: Evidence suggests an
alternative gene structure, and another ab
initio gene model PARTIALLY confirms it.
Target with
red check mark: Evidence suggests an
alternative gene structure, and another ab
initio gene model PARTIALLY confirms it.
 Target with
green check mark: Evidence suggests an
alternative gene structure, and a yrGATE
community annotation gene model PARTIALLY
confirms it.
Target with
green check mark: Evidence suggests an
alternative gene structure, and a yrGATE
community annotation gene model PARTIALLY
confirms it.
 Target
with red and green check marks: Evidence
suggests an alternative gene structure, and
another ab initio gene model AND a
yrGATE gene model each PARTIALLY confirm it.
Target
with red and green check marks: Evidence
suggests an alternative gene structure, and
another ab initio gene model AND a
yrGATE gene model each PARTIALLY confirm it.
No annotation action is warranted for the following types:
 Red check mark
with no target: Evidence suggests an
alternative gene structure, and another ab
initio gene model COMPLETELY confirms it.
Red check mark
with no target: Evidence suggests an
alternative gene structure, and another ab
initio gene model COMPLETELY confirms it.
 Green check
mark with no target: Evidence suggests an
alternative gene structure, and a yrGATE gene
model COMPLETELY confirms it.
Green check
mark with no target: Evidence suggests an
alternative gene structure, and a yrGATE gene
model COMPLETELY confirms it.
 Red and
green check marks with no target: Evidence
suggests an alternative gene structure, and
another ab initio gene model AND a
yrGATE gene model each COMPLETELY confirm it.
Red and
green check marks with no target: Evidence
suggests an alternative gene structure, and
another ab initio gene model AND a
yrGATE gene model each COMPLETELY confirm it.
References
yrGATE Tool:
Wilkerson, M.D., Schlueter, S.D. & Brendel, V. (2006) yrGATE: a web-based gene-structure annotation tool for the identification and dissemination of eukaryotic genes. Genome Biol. 7, R58. [ PubMed ID: 16859520 ]
Annotation Methods:
Brendel, V. (2007) Gene structure annotation at PlantGDB. In D. Edwards (ed.), Plant Bioinformatics: Methods and Protocols. Methods in Molecular Biology 406, 519-531. Humana Press, Totowa, NJ. Publication date: August 2007. [ PubMed ID: 18287710 ] | [ Publisher's Website]
Genome Browsers:
Schlueter S.D., Wilkerson M.D., Dong Q., & Brendel V. (2006) xGDB: open-source computational infrastructure for the integrated evaluation and analysis of genome features. Genome Biol. 7(11): R111. [ PubMed ID: 17116260 ] | [ online article ]
Cognate Loci in Arabidopsis:
W. Zhu, W., Schlueter, S.D., and Brendel, V. (2003) Refined annotation of the Arabidopsis thaliana genome by complete EST mapping. Plant Physiol. 132: 469-484. [ [PubMed ID: 12805580] | [ online article]