


|
Alternative splicing visualization tool...
|
Sources of New AS Events Identified in ASIP
One anonymous reviewer's suggestion was to apply our methodology to two previously published data sets on which other authors reported much fewer cases of alternative splicing. This control study should discriminate and quantify two contributing factors to the higher incidence rates of alternative splicing we found -- (1) possibly more accurate methods for determining evidence for alternative splicing, and (2) use of more comprehensive EST and cDNA collections now available.
We downloaded the TIGR transcript data set (208,604 sequences) used in the Haas et al. study (1)from TIGR�s website (http://www.tigr.org/tdb/e2k1/ath1/pasa_annot_updates/ ESTs_n_cDNAs.tar.gz), applied the same methodology described in our manuscript to this set, and found a total of 2,053 Arabidopsis annotated genes to be alternatively spliced (compared to only 909 genes identified by Haas et al.). More precisely, as shown in Figure A below, the overlap with TIGR�s AS gene list consists of 822 genes also identified in our comprehensive study (ASIP), 16 genes found by our methods only in our control study with the TIGR transcript set, 22 genes identified in our comprehensive ASIP study but not in the control study, and 49 genes were not identified by our method with either transcript data set. Assuming TIGR�s determination was entirely correct, the false negative rate of our method would be estimated in the range 2.4% -- 7.8% (22 -- 71 / 909). However, as there are clearly false positive determinations in the set of 49 missed genes, our actual false negative rate is certainly closer to the lower bound of the range (see manuscript Page 8 and Supporting Table 5).
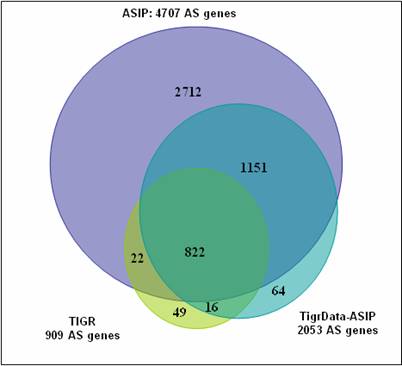 Figure A. Comparison among alternatively spliced (AS) genes identified in the TIGR study (1), our large-scale study (ASIP) and our control study on the TIGR data set (ControlStudy).
Figure A. Comparison among alternatively spliced (AS) genes identified in the TIGR study (1), our large-scale study (ASIP) and our control study on the TIGR data set (ControlStudy).
The 64 genes identified as alternatively spliced in our control study but not identified by either TIGR in their study nor in our comprehensive ASIP study provide a rough estimate of ~3.1% (64/2053) for the false positive rate. Closer inspection suggests that not all of these cases are false positive. Some of these events are reliable AS events missed in the comprehensive study, and some involve the small set of 418 transcripts in the TIGR transcript set that were not included in our ASIP set.
Some proportion of false determination is unavoidable for a large-scale study because of the uncertainties for some spliced alignments. Manual inspection of the problematic cases show that they almost all involve very short exon segments, less than perfect sequence matching, wrong strand alignments, or non-canonical splice sites. A number of these cases could be unambiguously resolved and have been corrected in our database. Our conclusions remain robust given the estimated error margins.
Surprisingly, we identified 1,151 alternatively spliced genes in our control study with the TIGR transcript set that were not reported by Haas et al (1). We cannot exclude the possibility of some false positive cases in this set, however the vast majority of these events are reliably identified and represent the AS events that were either missed or filtered by TIGR. In fact, many of these AS events have been incorporated in the current release of the Arabidopsis genome annotation.
In summary, our comprehensive ASIP study identified an additional 3,863 AS genes relative to the TIGR study. Based on the control study suggested by the reviewer, we estimate that 1,151 (30%) of these were contributed by the increased sensitivity of our method, and that the remaining 2,712 (70%) were contributed by the larger cDNA/EST data set used.
By applying our methods to the 280,569 RAFL clones available at RIKEN website (ftp://pfgweb.gsc.riken.jp/rafl/sequence/), AS events were identified in a total of 1,708 annotated genes, of which 1,093 appear also in the list of 1,348 alternatively spliced genes reported by Iida et al. based on BLAST alignments(2). 1,314 genes overlap with our ASIP comprehensive study. Of the 615 genes not identified by Iida et al., 394 (~64%) are of AltD, AltA, and AltP types. Of the 255 genes on Iida et al.�s list of alternatively spliced genes not found in our control study, 89 are of the AltD or AltA types. These differences seem to result from the differences in the methods with respect to splice site identification, which is explicitly modeled in GeneSeqer but not taken into account in BLAST. 107 of the 255 genes missed in our control study are annotated by RIKEN as ExonS events. Most of them involve single-exon transcripts or terminal exons, which were intentionally filtered out in our study (see Materials and Methods).
Links
- 1,151 alternatively spliced genes in our control study but not in TIGR's study.
- 64 possible false positive alternatively spliced genes in our control study using TIGR's dataset.
- 16 alternatively spliced genes in our control study and TIGR's study but missed in ASIP comprehensive study
- 615 alternatively spliced genes in our control study using RIKEN's dataset, but missed in RIKEN's study
- 255 alternatively spliced genes reported in RIKEN's study but missed in our control study.
References
- Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K., Jr., Hannick, L. I., Maiti, R., Ronning, C. M., Rusch, D. B., Town, C. D., Salzberg, S. L. & White, O. (2003) Nucleic Acids Res 31, 5654-5666.
- Iida, K., Seki, M., Sakurai, T., Satou, M., Akiyama, K., Toyoda, T., Konagaya, A. & Shinozaki, K. (2004) Nucleic Acids Res 32, 5096-5103.
|
 |